
In eukayotic organisms, DNA is organized into repeating units called nucleosomes, each of which consist of ~1.65 turns of DNA wrapped around an octamer of histone proteins. The array of nucleosomes is folded into a thicker chromatin fiber that undergoes further looping to eventually yield chromosomes. Our group is interested in developing novel computational methods to elucidate this 3D organization of DNA and its role in biological processes like transcription. Our earliest efforts involved the development of a state-of-the-art mesoscopic model of nucleosome arrays (Fig. 1) and a tailored Monte Carlo approach for simulating conformations of the arrays. Our modeling provided the first evidence of the chromatin fiber exhibiting a polymorphic structure, displaying a mixture of zigzag and solenoidal conformations; These results were verified by a EM-assisted nucleosome interaction capture experiments from the S. Grigoryev lab. The model also importantly allowed us to dissect the roles of the linker histone, salt condition, and the four types of histone tails in chromatin folding.
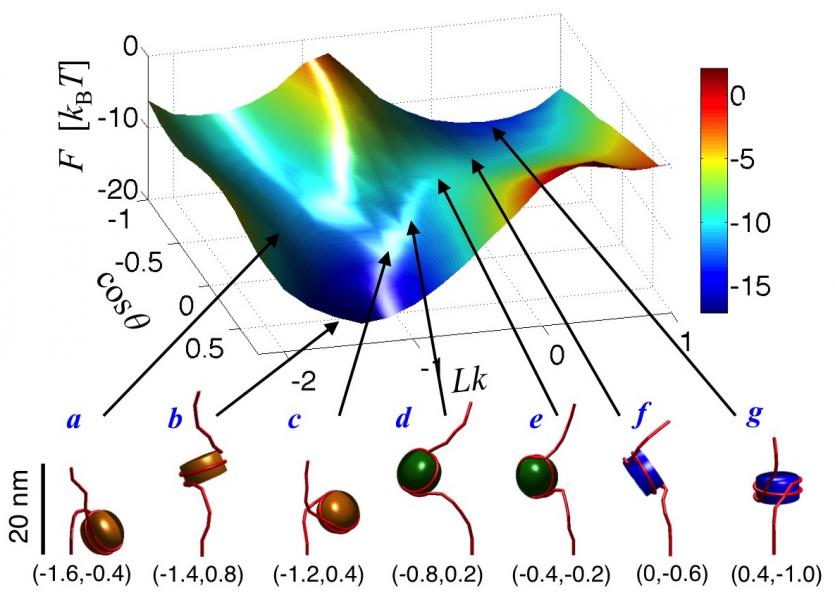
More recently, we have used these models to investigate the effects of torsional stresses on chromatin. DNA is subjected to myriad torsional stresses in vivo, which until now were viewed as a byproduct of biological processes that needed to be eliminated by specialized enzymes. However, new evidence suggests that torsional stresses serve important roles in gene regulation, but little is known about how these stresses propagate within chromatin and affect its organization. Our work now provides the first detailed picture of the structure and dynamics of torsionally stressed chromatin and revealed a new mechanism, nucleosome phasing, by which the torsional response of chromatin could be modulated. In related work, we have mapped out the free energy landscape of mononucleosomes (Fig. 2) and nucleosome arrays to investigate differences in the conformational fluctuations of nucleosomes in isolation and those present within arrays.

Another focus is on developing methods to elucidate the 3D organization of chromatin within chromosomal domains. We have developed a novel strategy for recovering ensembles of chromatin conformations from contact probabilities (CPs) between genomic loci, as measured from chromosome conformation capture experiments. Our approach involves iterative adjustment of parameters of a polymer model of chromatin via an adaptive filter approach until the CPs estimated from simulations of the model match those obtained experimentally (Fig. 3). To speed up ensemble recovery, we devised a new method for estimating CPs based on functional approximations of inter-bed distance distributions obtained from polymer simulations. We are currently using these methods to investigate structural changes in specific loci in association with B-cell development and cancer therapy, in collaboration with Dr. C. Murre and Dr. M. G. Rosenfeld. Concurrently, we are developing software that will allow researchers to obtain enzyme cleavage fractions in their Hi-C experiments and obtain CP maps with much higher resolution than afforded by current methods.