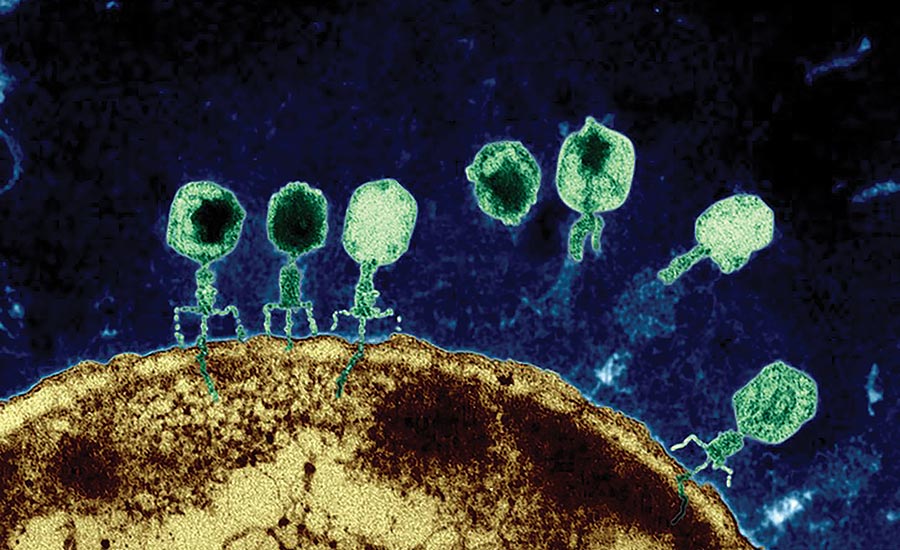
Many viruses use powerful molecular motors to package their genome into procapsids during assembly (Fig. 1). These motors are collectively known as packaging ATPases, which convert the chemical energy from ATP hydrolysis into mechanical work. These motors can generate force in excess of 60 pN, and package DNA at rates up to 2000 bp/s. Thus, they represent the most powerful molecular motors known to mankind. We are broadly interested in the mechanisms by which these proteins function. Uncovering such mechanisms would not resolve a fundamental problem in virology but also provide insights into combating viral infections like herpes and designing synthetic mimics of these powerful motors.

Our initial foray into the field was testing a previously proposed mechanism of packaging for the T4 bacteriophage, which suggested that an extended-to-compact transition of the protein mediated by electrostatic interactions between complimentarily charged residues generated the force needed to package DNA. In collaboration with Douglas Smith (UCSD) and Venigalla Rao (Catholic U America), we showed that site-directed alterations in these residues cause force dependent impairments of motor function that correlate well with computed changes in free-energy differences between the two states, thus providing support for the proposed model. Using free energy decomposition analysis, we were able to identify all the key molecular interactions and residues involved in force generation, revealing that uncharged residues also play an equally important role in compaction. We also proposed an energy landscape model of motor activity under external loads that couples the free-energy profile of motor conformational states with that of the ATP hydrolysis cycle (Fig. 2).
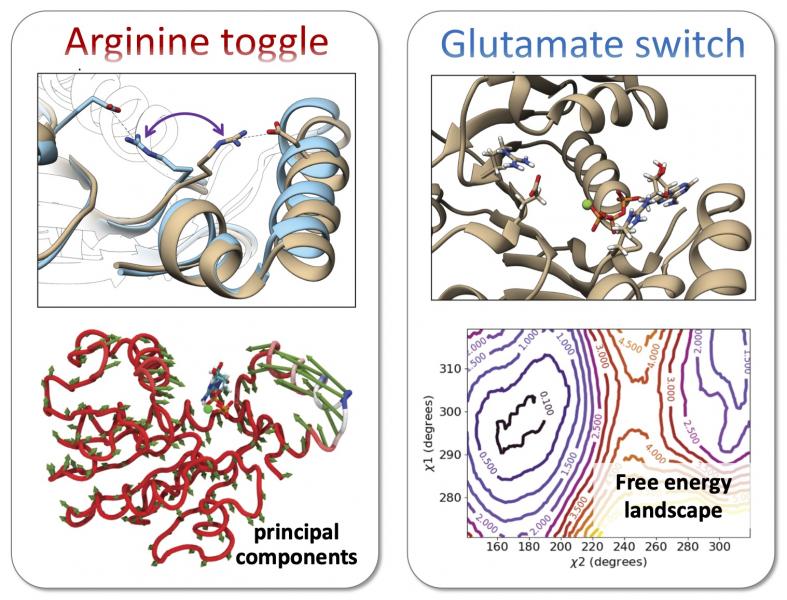
Our more recent work has attempted to model the packaging process at multiple scales. At the finest scale, we have used atomistic simulations to identify mechanisms involved in mechanochemical coupling and ATP hydrolysis (Fig. 3). Simulations of the T4, Sf6, and P74-26 phage packaging ATPases ran on the NSF XSEDE compute cluster predicted that a highly conserved “arginine toggle” couples nucleotide occupancy to lid subdomain rotation, consistent with experimental data collected by our collaborators: Douglas Smith (UCSD), Carlos Catalano (UC Denver), and Michael Feiss (U Iowa). In a follow-up study, we provided atomistic insight to explain functional roles of the Walker B motif of these packaging ATPases. We are currently using 2D umbrella sampling simulations and weighted histogram analysis methods methods to interrogate whether packaging ATPases use a glutamate switch mechanism commonly found in AAA+ ATPases to modulate the rate of ATP hydrolysis in order to coordinate catalytic events across subunits.
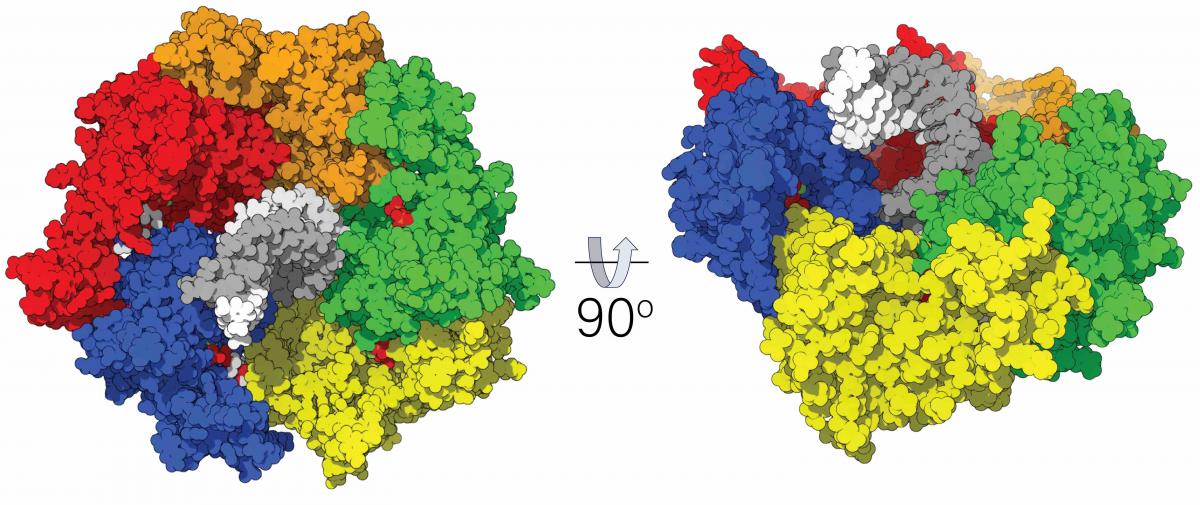
At a larger scale, we are utilizing the immense power of the Anton2 supercomputer to perform first-of-their-kind simulations of pentameric packaging ATPase complexes (Fig. 4). Our simulations of the P74-26 and D6E phage packaging ATPases predict that these pentameric structures undergo a planar-to-spiral transition to accommodate the helicity of double-stranded DNA. These spiral rings are consistent with recently solved cryo-electron microscopy structures of hexameric AAA+ motors, such as Vps4p and katanin, which also adopt spiral conformations when engaged with their substrate. We are currently using approaches like nudged elastic band method to sample the minimum-energy pathway connecting the observed states. We are also working closely with the experimental labs of Marc Morais (UTMB) and Paul Jardine (U Minnesota) to compare our modeling predictions with structural and biochemical data from the phi28 and phi29 bacteriophage systems.

At the largest scale, we are combining atomistic and coarse-grained simulations to study the dynamics of DNA as it is packaged into the procapsid. We are focusing on how environmental conditions within the host bacteria, such as multivalent salt concentration, can affect how DNA interacts with the capsid surface, and thus alter the conformation of the packaged genome (Fig. 5).